Development of a simulation platform for multi-layered electrochemical cells
1. Motivation
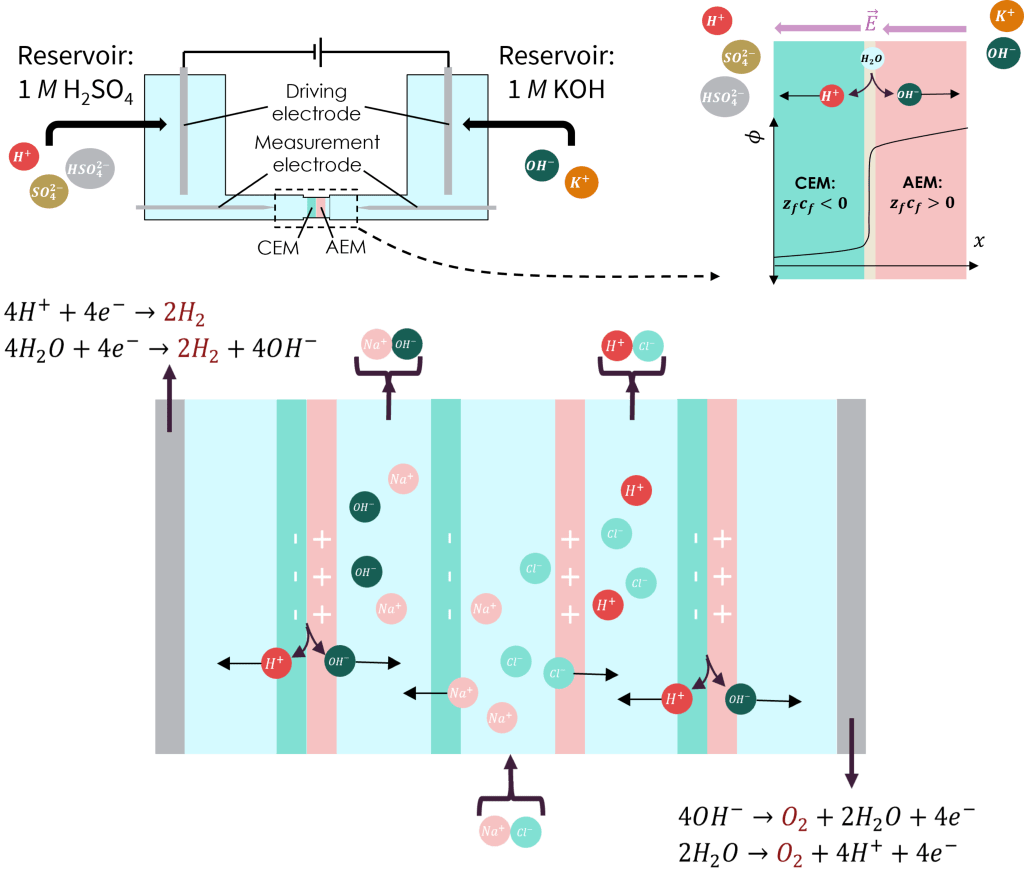
Electrochemical cells with varying applications and operating modes may be constructed by layering materials with different physical and chemical properties in specific arrangements. Consider, for example, bi-polar membranes, which are constructed by lamination of an anion- and cation-selective membrane. Thinking more broadly, an entire electrochemical cell (i.e., the cell shown above, which produces acids/bases from salts) may be represented in 1D as a multi-layered structure comprising anion/cation-selective membranes and electrodes separated by bulk diffusion layers.
The purpose of this project is development of a general-use solver for multi-layered electrochemical cells. The physical, transport, and chemical-kinetic properties of each “layer” are fully user-specifiable via a simple input file, allowing researchers to construct simulation domains with arbitrary arrangements of electrochemical components and to run efficient 1D simulations for a wide variety of applications without writing new code.
2. Membrane Chemistry

Each “membrane” layer is characterized geometrically by its porosity and tortuosity, parameters that describe the volume-ratio and morphology of void regions within the porous polymer structure. Ionic surface groups are responsible for the perm-selectivity of each membrane, which may be captured in the simplest way by setting a fixed “background charge density” attributable to those surface groups.
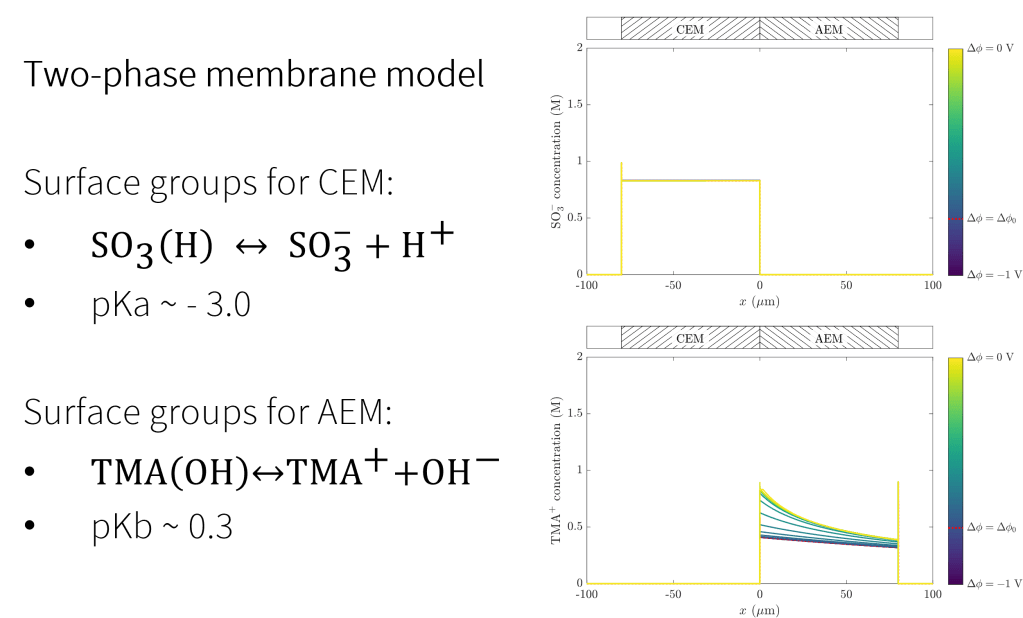
Our simulation platform permits study of cases in which extreme variation of pH may require consideration of surface reactions involving ionic groups in the membranes. Shown above are concentration profiles across a bi-polar membrane for sulfonic groups and TMA+, functional groups commonly responsible for selectivity of cation- and anion-selective membranes, respectively. For the range of cell voltages shown here, the strongly-acidic sulfonic acid groups remain de-protonated, while the TMA(OH) groups encounter a pH swing large enough to alter the effective “background charge density.”
3. Bipolar Junction Chemistry

Recent investigations have shed light on the causes of enhanced water dissociation kinetics at the junction region. Our platform allows users to incorporate the Second Wien Effect, whereby the presence of a strong electric field shifts the effective dissociation rate coefficient for polarizable molecules.
4. Numerical Methods
We offer a brief summary of the key numerical methods; an exhaustive description will appear in an upcoming publication. Second-order central differences are used for spatial discretization, and a third-order, implicit, multi-step method is used for temporal discretization. We incorporate adaptive time-step refinement, permitting resolution of rapid initial transients despite selection of a large time-step to reach steady-state results. Similarly, the mesh generation module permits exponential mesh refinement near interfaces between layers, where steep concentration and potential gradients are expected. This combination of methods permits full-cell simulations over long time-scales, granting users the ability to time-advance until reaching steady-state.
5. Notable Results
A thorough description of the simulation platform and analysis of notable findings for bipolar membranes under forward-biased electric potentials is the focus of an upcoming publication, with expected submission in mid to late 2025.
Acknowledgements
This work was supported by the Solar Photochemistry and Catalysis Programs of the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, U.S. Department of Energy, under Grant No. DE-SC0021633. Simulations were performed on the Armstrong and Shepard clusters at the Stanford High Performance Computing Center.
